ЭПИЛЕПСИЯ• Главная страница
• Эпилепсия в вопросах и ответах
• Конференция
Для специалистов• Библиотека эпилептолога• Библиотека невролога • Новости эпилептологии • Антиконвульсанты |
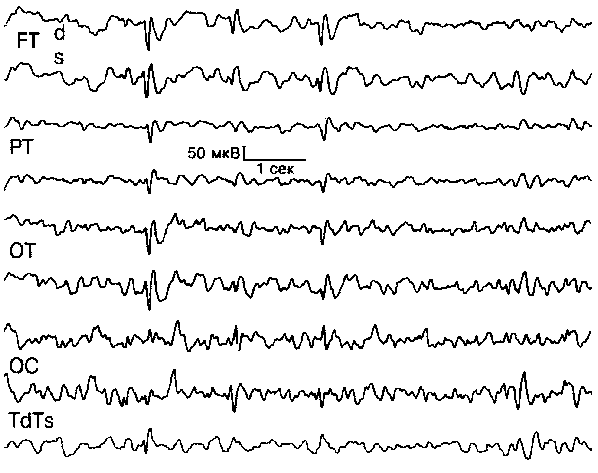
Роландическая эпилепсияК.Ю. Мухин, П.А. Темин, Е.А. РыковаRolandic epilepsy Кафедра нервных болезней педиатрическогофакультета (и.о. зав - профессор Л.В. Калинина)Российского Государственного медицинскогоУниверситета Луиджо Роландо (1773-1831 г.), итальянский врач ианатом, дал классическое топографическоеописание центральной борозды коры головногомозга. При идиопатической парциальной эпилепсиидетского возраста, характеризующейся короткимигемифациальными моторными ночными приступами,часто с предшествующей соматосенсорной аурой итипичными изменениями ЭЭГ (полифазные спайки слокализацией в центральной и срединной височнойобластях) [14], эпилептогенной зоной являютсянижние отделы роландической борозды. Изучениеэтой разновидности идиопатической парциальнойэпилепсии начато более 40 лет назад.Первоначально были описаны специфическиеизменения ЭЭГ у определенной группы детей. В 1952 г.H.Gastaut [25] впервые обратил внимание на особыеэпилептические изменения ЭЭГ у некоторых детей:полифазные спайки со строгой локализацией вперироландической области. Первое клиническоеописание симптомов эпилепсии с роландическимиспайками (1958 г.) принадлежит Р.Nayras и М.Веаussart [40]. Впоследующем Е.Gibbs и F.Gibbs [27], С.Lombroso [36], Р.Loisea исоавт. [35] провели клинический анализ симптомовпри эпилепсии с роландическими спайками ипредложили называть данную форму эпилепсиироландической. Авторы отметили ее благоприятныйпрогноз и отсутствие симптомов органическогопоражения центральной нервной системы. Частота Роландическая эпилепсия (РЭ) - одна из наиболеечастых форм эпилепсии детского возраста.Распространенность ее составляет 21 на 100 тыс.здорового детского населения [29]. Частота РЭсреди всех форм эпилепсии с дебютом до 13 летварьирует по разным данным от 11,5% [15] до 25% [5]. Из 360больных эпилепсией детей, наблюдавшихся Р.Lerman [34],у 14,4% была диагностирована РЭ. Для сравнениязаметим, что одна из наиболее частых формэпилепсии детского возраста - пикнолепсия - быладиагностирована лишь у 10,5% пациентов. Неисключено, что истинная частота РЭ в популяциизначительно выше, так как у многих больныхотмечаются единичные ночные приступы, частоостающиеся без внимания родителей и врачей [34]. Большинство авторов отмечают преобладаниемальчиков среди больных РЭ [3]. Соотношениемальчиков и девочек в среднем составляет 6:4 [11].Среди 100 бальных, наблюдавшихся Р.Lerman [34], было 62мальчика и 38 девочек. Клиника Дебют РЭ варьирует в возрастном интервале 2 - 14лет. В 83% случаев РЭ дебютирует в возрасте 4 - 10 лет[5]. У подавляющего большинства пациентовприступы начинаются между 5-м и 10-м годом жизни смаксимумом в возрасте 9 лет [9]. Средний возрастпоявления приступов - 9,9 года [33]. Началозаболевания в возрасте до 2 лет отмечается нечаще чем в 8% случаев [15]. Имеются единичныеописания начала РЭ на 1-м году жизни [17], однакоотнесение данных случаев к РЭ дискутируется [21].Начало заболевания после 11 лет также являетсяредкостью, а после 14 лет такие случаи ненаблюдаются [9]. Клиническая симптоматика приступов при РЭ, какправило, типична. Наблюдаются простыепарциальные (моторные, сенсорные, вегетативные),сложные парциальные (моторные) и вторичногенерализованныс приступы. Наиболее типичныпростые парциальные моторные и/или сенсорныепароксизмы [2]. Простые парциальные приступы составляют"ядро" РЭ и наблюдаются у 70 - 80% больных.Наиболее типично начало приступа ссоматосенсорной ауры: ощущение покалывания,онемения, "прохождения электрического тока"унилатерально в области глотки, языка, десны.Вслед за аурой развивается парциальный приступ.Возможны следующие варианты: гемифациальныеприступы; унилатеральные тонические,клонические или тонико-клонические судорогимышц лица; фарингооральные приступы;унилатеральные судороги губы, языка, глотки,гортани, часто сочетающиеся с анартрией игиперсаливацией. Гемифациальные приступы имеютместо у 37% больных, фарингооральные - у 53% [91. Частово сне больные издают своеобразные горловыезвуки типа "бульканья", "хрюканья","полоскания горла". Истинную частотусоматосенсорных приступов (щечная парестезия потерминологии J.Aicardi [5]) определить сложно, так какдети, особенно маленькие, затрудняются описатьсвои ощущения во время приступов. У 20% больных судороги могут распространяться смышц лица на гомолатеральную руку(брахиофациальные приступы) и примерно в 8%случаев вовлекать и ногу (унилатеральныеприступы). По мере развития заболевания приступымогут менять сторонность. Вторичногенерализованные судорожные приступыотмечаются у 20-25% больных РЭ. Они наиболеехарактерны для младших детей, и возникновениегенeрализованных приступов "жестко"приурочено ко сну. Наличие у пациентовдлительное время только генера- лизованныхсудорожных приступов нехарактерно для РЭ, как иналичие абсансов, психомоторных приступов иприступов с психическими симптомами [5]. Продолжительность приступов при РЭ, какправило, небольшая [5] от нескольких-секунд до 2-3мин. У 11% [16] - 22% [5] больных продолжительностьпароксизмов превышает 10-15 мин, у 16% [5] отмечаютсятяжелые продолжительные приступы,заканчивающиеся постприступным гемипарезом(паралич Тодда). Частота приступов при РЭ обычноневысока - в среднем 2 в год [5]. В первые 1-2 года смомента дебюта заболевания приступы могут бытьболее частыми, однако с течением времени онивозникают все реже. После 13-16 лет возможно полноеисчезновение приступов. У 10-40% детей наблюдаетсяединичный приступ за все время болезни, даже еслиони не получают антиконвульсантов [41].Еженедельные и более частые, ежедневные приступынаблюдаются лишь в 6% [35] - 20% [34] случаев. Серийныеприступы и повторные в течение одной ночи редки инаблюдались в 3% случаев, описанных Т.Deonna и соавт.[17]. В редких публикациях [15,16,23,34] сообщается оразвитии эпилептического статуса при РЭ. Частотаего при РЭ составляет около 11% [16]. Подробноеописание эпилептического статуса у больных РЭпредставлено в работе N.Fejerman и А.Di Blasi [23]. Авторынаблюдали 2 мальчиков 8 лет с дебютом РЭ в 3 года, укоторых возник длительный статус гемифациальныхприступов. Сознание было сохранено, однакоконстатировались анартрия и выраженноеслюнотечение. Пероральное назначениеантиконвульсантов в высоких дозах, как ипарентеральное введение диазепама илилоразепама, не оказало влияния на эпилептическийстатус. Лишь назначение гормональных препаратов(дексаметазон) полностью купировало судороги.Вызывает сомнение, однако, отнесение данныхслучаев к РЭ. У одного из этих пациентовнаблюдались типичные абсансы, у другого -приступы с джексоновским маршем; у обоихприступы были резистeнтны к антиконвульсантам.Возможно, данные случаи представляют собойатипичную форму РЭ. Е.Roulet и соавт. [42] описалиэпилептический статус у мальчика 6 лет с РЭ.Единственным его проявлением было длительноеслюнотечение в сочетании с оролингвомоторнойдиспраксией. На ЭЭГ во время слюнотечениярегистрировались спайк-волновые комплексы,включая роландические. При увеличении дозыкарбамазепина слюнотечение прекратилось. Помнению авторов, длительное перманентноеслюнотечение у больных РЭ может бытьединственным симптомом эпилептического статуса. Пароксизмы РЭ связаны с ритмом сон -бодрствование. Наиболее типичны ночные приступы,возникающие преимущественно при засыпании ипробуждении. При ночных приступах пароксизмывозникают в период пробуждения (35%), реже - всередине ночи (25%) и при засыпании (20%). У 25-30%больных приступы наблюдаются как во сне, так и всостоянии бодрствования [5]. Лишь у 5-25% пациентовони возникают исключительно со времябодрствования [5,34]. Имеется некоторое различиекартины приступов в зависимости от ритма сон -бодрствование. Дневные приступы практическивсегда простые парциальные, чаще гемифациальные,короткие, ночные, как правило, более тяжелые,продолжительные, обычно унилатеральные иливторично генерализованные. Провести оценкусознания у пациентов с ночными приступамисложно; феномен анартрии часто симулируетклинику выключения сознания. Сложныепарциальные приступы при РЭ редки и составляютлишь около 5% всех случаев [15]. Возможно, напротяжении приступа у некоторых больных имеетместо "флюктуация" уровня нарушениясознания. J.Aicatdi и J.Сhevrie [4] была выделена группа больных сосвоеобразным эпилептическим синдромом,клиническая симптоматика которого была схожа сРЭ. У больных отмечались простые парциальныегемифациальные и гемиклони-ческие приступы,однако в сочетании смиоклонически-астатическими, атоническимиприступами и в ряде случаев с абсансами. Даннаягруппа наблюдений классифицирована какатипичный вариант РЭ. Частота его, согласно Т.Deonnaи соавт. [16], составляет 5% среди всех больных РЭ.Таксономическое положение данного синдромаспорно. По мнению J.Aicatdi и J.Сhevrie [5], данная формаэпилепсии является нозологическисамостоятельным синдромом с четко очерченнымиклиникой, изменениями ЭЭГ и прогнозом. Другиеавторы предполагают наличие атипичного вариантас рамках РЭ [17]. Подробное описание данногосиндрома представлено Т.Deonna и соавт. [16] напримере 6 пациентов. Среди обследованных больныхпреобладали мальчики. Приступы дебютировали ввозрастном интервале от 2 до 7 лет уневрологически нормальных детей с сохранныминтеллектом. Клинические симптомы включалиразличные виды приступов. Характерными былиночные простые парциальные приступы, подобныетаковым при РЭ, миоклонически-астатические иатонические приступы. У ряда больных отмечалисьатипичные абсансы. Приступы во всех случаях быличастыми, ежедневными и приводили к многократнымпадениям пациентов и тяжелым ушибам. На ЭЭГтипичные роландические спайки сочетались смедленными комплексами пик-волна, характернымидля синдрома Леннокса - Гасто. Отмечалосьнарастание эпилептической активности в фаземедленного сна. Клиническая симптоматика атипичной РЭ носитчерты целого ряда эпилептических синдромов: РЭ(простые парциальные ночные приступы в сочетаниис типичными роландическими спайками), синдромаЛеннокса - Гасто (частые атонически-астатическиеприступы, атипичные абсансы в сочетании смедленными комплексами пик-волна) и эпилепсии смиоклонически-астатическими приступами,описанной Н.Doose [19]. Однако кардинальным отличиематипичной РЭ от синдрома Леннокса - Гасто иэпилепсии с миоклоничeски-астатическимиприступами являются отсутствие у пациентовнарушений интеллекта и хороший прогноз. Р.Loisea исоавт. [35] провели катамнестическое исследование13 пациентов с атипичной РЭ в течение не менее 9лет. Спустя разный промежуток времени во всехслучаях была достигнута полная ремиссия, приэтом большая часть пациентов (10) не применялирегулярной противоэпилептической терапии.Авторы не отметили ни отставания в психическомразвитии, ни серьезных нарушений поведения как внаиболее активный период заболевания, так и впериод ремиссии, что сближает данные случаи стипичной РЭ. По мнению авторов, при данномсиндроме следует избегать "агрессивной"противоэпилептической терапии ввиду хорошегопрогноза. O.Dulac и соавт. [21] проанализировали случаипарциальной эпилепсии с ранним дебютом - между 8днями и 3 годами жизни. Среди 442 больных выявлено 17с парциальной эпилепсией с ранним дебютом.Приступы характеризовались как простыепарциальные с частой вторичной генерализацией.По мнению авторов, данные случаи являютсяпереходной формой между доброкачественнойэпилепсией новорожденных и РЭ. В отличие от двухуказанных синдромов прогноз был неблагоприятными приступы не поддавались медикаментознойтерапии. Последующие исследования покажут,являются ли случаи парциальной эпилепсии сдебютом в первые 3 года жизни нозологическисамостоятельным синдромом или еще одним"атипичным вариантом" РЭ. Неврологический статус Неврологический статус у детей, страдающих РЭ,изучен достаточно подробно. РЭ относится кидиопатическим формам эпилепсии, при которыхотсутствуют признаки органического пораженияголовного мозга, что подтверждаетсябольшинством публикаций. Более того, по мнениюА.Веаumanoir и соавт. [8]. наличие таких симптомовпротиворечит критериям диагноза РЭ. Однако впоследние годы благодаря внедрению вклиническую практику высокоэффективныхнейрорадиологических методов исследования(компьютерная томография, ядерно-магнитныйрезонанс, позитронная эмиссионная томография)появились единичные сообщения об обнаруженииструктурных изменений в головном мозге у больныхРЭ. Из 100 больных, наблюдавшихся Р.Lerman и S.Kivity [33], у 4констатированы симптомы органическогопоражения центральной нервной системы: детскийцеребральный паралич (гемипаретическая итетраплегическая формы), микроцефалия, умереннаязадержка умственного развития. Во всех этихслучаях диагноз РЭ не вызывал сомнения и былподтвержден как клинически, так инейрофизиологически. Пневмоэнцефалография невыявила нарушений у данных больных. S.Blom и J.Heijbet [11]в 3 (7,5%) из 40 случаев РЭ обнаружили у больныхцентральный гемипарез. E.Roulet и соавт. [42] отметилипроявления оролингвомоторной диспраксии убольного РЭ. Такие симптомы, как микроцефалия,косоглазие, мозжечковая недостаточность,задержка психического и речевого развития,констатировали у ряда больных РЭ Р.Santanelli и соавт.[43]. Синдром гиперактивности был обнаружен у 16,7%больных РЭ [34]. Р.Вrау и W.Wiser [13] первыми констатировали наличиесемейных случаев РЭ и предположилиаутосомно-доминантную модель наследования снеполной пенетрантностью и возрастнойзависимостью. В последние десятилетия проведенымногочисленные клинико-генеалогическиеисследования при РЭ, подтверждающиеправомерность данной концепции. Согласнообобщенным данным литературы, 8,9% [35] - 68% [29]больных РЭ имеют родственников, страдающихэпилепсией или имеющих приступы в анамнезе, и до30% [13] - родственников с наличием на ЭЭГроландических спайков при отсутствии приступов.Частота обнаружения спайков у сибсов пробандов идетей, родители которых страдали РЭ, составляет25-36% (в популяции здорового населения - 1,4-5% [З7]).Однако лишь у малой части из них (12% висследовании Р.Вrау и W.Wiser [13]) отмечалисьприступы: остальные были клинически здоровы.Характер приступов у родственников больных РЭможет быть различным. Типичные приступы,характерные для РЭ, констатируются у 13%родственников, абсансная эпилепсия - у 10%,гснсрализованные судорожные приступы - у 3%,фокальная эпилепсия - у 1% [9]. В ряде исследований изучена конкордантностьмонозиготных близнецов по РЭ. Обнаруженавысочайшая конкордантность по наличию на ЭЭГроландических спайков, но не по развитию самойРЭ. T.Kajitani и соавт. [30] изучили 3 пары монозиготныхблизнецов в возрасте 5-11 лет, в которых один изсибсов страдал РЭ. Во всех случаяхконстатирована типичная роландическаяэпилептическая активность на ЭЭГ, но ни у одногоиз здоровых сибсов не было никаких проявлений РЭ. В последние годы была выдвинута гипотеза омульти-факторном наследовании РЭ [29]. Онаосновывается на том факте, что лишь у небольшойчасти детей, имеющих на ЭЭГ роландические спайки,в дальнейшем развивается РЭ. Предполагается, чтороландические спайки и развитие РЭдетерминированы двумя различными, носцепленными генами. В 1982 г. O.Eeg-Olofsson и соавт. [22]обнаружили, что у родителей, сибсов и другихродственников детей, больных РЭ, низкаявстречаемость гаплотипа А1В8 антигеновлейкоцитарной гистосовместимости. Частота его вобщей популяции крайне высока [22]- Возможно, чтоданный гаплотип играет роль протектора илиингибирующего фактора, препятствующего развитиюРЭ. Интересны исследования, раскрывающиевзаимосвязь РЭ с фебрильными судорогами. E.Frantzen исоавт. [24] сообщили, что у 20% детей с фебрильнымисудорогами в дальнейшем на ЭЭГ появляетсяфокальная эпилептическая активность, идентичнаяроландическим спайкам. Ни у одного из этихпациентов не отмечалось в дальнейшемэпилептических приступов. Исследования намонозиготных близнецах также показали возможноегенетическое сцепление между РЭ и фебрильнымисудорогами. M.Lennox-Buchthul [32] изучила 24 парымонозиготных близнецов, в которых по крайнеймере у 1 из пары имелись фебрильные судороги, ивыявила, что 20% их сибсов имели редкие ночныеприступы. У 2 из 24 обнаружено сочетание ночныхприступов с фебрильными судорогами. К сожалению,автор не идентифицировала данные ночныеприступы, однако по характеру их течения ивозрасту дебюта можно предположить, что этослучаи РЭ. В исследовании T.Kajitani и соавт. [31] среди 3пар монозиготных близнецов 3 имели РЭ и 5 -фебрильные судороги. Данные работы могутсвидетельствовать о генетическом сцеплении РЭ ифебрильных судорог. Подтверждением этойгипотезы служит и клиническая схожесть двухданных синдромов: высокая генетическаядетерминированность, "жесткий"возрастзависимый дебют, доброкачественныйпрогноз [1] . Остается неясной роль экзогенных, средовыхфакторов в развитии РЭ. H.Doose и W.Baier [18]предположили, что генетические факторы при РЭдетерминируют низкий порог судорожнойготовности головного мозга. При этом здоровыхдетей, имеющих на ЭЭГ роландические спайки, онирасценили как субклинических носителей с низкимсудорожным порогом. По мнению авторов,воздействие различных экзогенных факторов вонтогенезе, а также сцепление с другими генамиможет способствовать у таких пациентовклинической манифестации заболевания иоказывать некоторое влияние на его течение.Т.Каjitani и соавт. [30] описали 3 родных сибсов, укаждого из которых на ЭЭГ имелись типичныероландические спайки. У одного из сибсов РЭдебютировала в 4 года во время лихорадочногозаболевания, у другого отмечался единственныйприступ в возрасте 10 лет при просмотретелепередач, третий миновал "критическийпериод" для дебюта и был здоров. Данноенаблюдение иллюстрирует полиморфизмклинических проявлений заболевания, что можетзависеть от различных экзогенных воздействий. Факторами, трансформирующими субклиническоеносительство в РЭ, могут быть патологиябеременности и родов, травмы головы,нейроинфекции и пр. [5]. Однако более чем уполовины детей, страдающих РЭ, указание наналичие данных факторов в анамнезе отсутствует[15]. Электроэнцефалография Необходимым исследованием для объективизациидиагноза РЭ является электроэнцефалография [14].На ЭЭГ в межприступном периоде при РЭобнаруживаются характерные, типичные"роландические" или "центровисочные"комплексы при обязательно сохранной основнойактивности. Эти комплексы представляют собоймедленные дифазные высокоамплитудные пики илиострые волны (100 - 300 мВ), нередко с последующимимедленными волнами, общей продолжительностьюоколо 30 мс (см. рисунок). Они имеют тенденцию квозникновению группами. Данные комплексынапоминают зубцы QRST ЭКГ. Роландические комплексы"жестко" локализованы: центральная ицентровисочная области. Роландические комплексымогут наблюдаться как унилатерально (обычноконтралатерально гемифациальным приступам) - 60%больных, так и билатерально - 40% [29].Эпилептические комплексы, как правило,независимы друг от друга, лишь в редких случаяхнаблюдается их билатерально-синхронноераспространение с амплитудным преобладанием содной стороны [26,34]. D.Gregory и Р.Wong [28] провели изучение точнойлокализации роландических спайков, используяметод компьютеризированного топографическогокартирования. Авторы обнаружили, что максимум"позитивности" электрического диполя при РЭнаходится в центрально-височной области, амаксимум "негативности" - в лобной.Предположено, что специфические ЭЭГ-паттерны приРЭ исходят из зоны, расположенной в нижнихотделах роландической борозды, на границе ссильвиевой бороздой. Типичной особенностьюизменений ЭЭГ при РЭ является нестойкостьпаттернов, их вариабельность от одной записи кдругой. Роландические спайки могут исчезать, азатем появляться вновь, менять стройность,конфигурацию даже при двух последующих записяхЭЭГ через короткий промежуток времени.Нестойкость ЭЭГ-паттернов при РЭсвидетельствует, по мнению J.Aicardi [5], об отсутствииорганических изменений в головном мозге при РЭ. Всвязи с этим отсутствие роландических спайковпри однократном злектро-энцефалографическомисследовании не может считаться убедительнымаргументом для исключения диагноза РЭ приналичии типичной картины приступов [15]. Крайневажным для подтверждения диагноза РЭ являетсяисследование ЭЭГ во время сна. Около 30% детей,страдающих РЭ, имеют роландические спайкиисключительно во время сна [29]. Во время дремоты ина всех стадиях сна отмечается тенденция кпереходу унилатеральных роландическихкомплексов в билатеральные [34]. Интересной находкой при РЭ являетсяобнаружение на ЭЭГ наряду с типичнымироландическими комплексами и другихэпилептических паттернов. От 10 до 20% детей с РЭимеют на ЭЭГ пик-волновые комплексы в другихзонах коры, главным образом в затылочной области[41]. Морфология "затылочных комплексов"близка к роландическим и к наблюдаемым придоброкачественной фокальной эпилепсии сзатылочными пароксизмами [10]. По мнению J.Aicardi [5],частота "затылочных комплексов" при РЭобратно пропорциональна возрасту ребенка иявляется нередкой у больных до 3 лет жизни. От 7% [34]до 20% [5] пациентов с РЭ демонстрируют на ЭЭГтипичную генерализованную пик-волновуюактивность с частотой 3-4 Гц, нередко возникающуюпри гипервентиляции. У отдельных больныхвозможно появление медленных пик-волновыхкомплексов. Большинство авторов придерживаютсяоднозначного мнения об отсутствии корреляциимежду частотой и выраженностью пик- волновыхкомплексов на ЭЭГ и особенностями течениязаболевания. Исследование ЭЭГ во время приступапредставляется довольно сложным ввиду низкойчастоты приступов при РЭ. B.Dalla-Bernardina и соавт. [15]описали во время ночного приступа появление наЭЭГ низкоамплитудной быстрой активности вцентрально-височной области, переходящей вроландические комплексы с распространением навсю гемисферу и с последующим охватом всегополушария. Нейрорадиологическое исследование Сведения о нeйрорадиологическом исследовании убольных РЭ единичны. H.Gastaut [25] одним из первыхизучил данные компьютерной томографии у 15больных РЭ и не нашел патологических измененийни в одном случае. В последующем принейрорадиологическом исследовании у больных РЭбыли обнаружены некоторые структурные измененияв головном мозге: расширение сильвиевой щели,наличие полости прозрачной перегородки (2 из 13случаев) [39], вентрикуломегалия, арахноидальныекисты (3 из 17 случаев) [38]. Р.Santanelli и соавт. [43]представили подробное клиническое инейрорадиологическое описание 3 случаев РЭ,сочетающейся со структурными изменениямиголовного мозга. У одного больного была выпиленалипома мозолистого тела, у другого (соспондилоторакальной дисплазией) - агенезиямозолистого тела с вентрикуломегалией, утретьего - множество церебральных,преимущественно перивентрикулярных,кальцификатов (врожденный токсоплазмоз). У всехэтих больных наблюдались типичная клиническаякартина РЭ и роландические спайки на ЭЭГ. Обнаруженные при нейрорадиологическомисследовании у единичных больных РЭорганические симптомы поражения головного мозгатрудно интерпретировать однозначно. Неисключено, что эти нарушения не имеютнепосредственной причинно-следственной связи сРЭ и могут быть вызваны сочетанием РЭ с другимизаболеваниями - детским церебральным параличом,микроцефалией, токсоплазмозом,эктомезодермальными дисплазиями. Как и впопуляции в целом, у больных РЭ могут наблюдатьсяперинатальное поражение центральной нервнойсистемы, постнатальные черепно-мозговые травмы.Частота перинатальных поражений центральнойнервной системы среди больных РЭ составляет 6% [16]- 13% [34], сотрясения головного мозга - 4 - 5% [34], что непревышает статистических данных в популяцииздоровых детей. Принципиально важный и до концане решенный вопрос - влияют ли данные экзогенныефакторы на возникновение и течение РЭ. По мнениюJ.Aicardi [5], нарушения, обнаруженные в головном мозгеу больных РЭ, не могут быть причиной эпилепсии, аявляются случайными находками. С другой стороны,нельзя игнорировать тот факт, что у ряда больныхРЭ структурные изменения с головном мозгелокализуются в нижних отделах роландическойборозды [43], а это не исключает полностьювозможность существования"симптоматической" формы эпилепсии. Лечение За последние годы в лечении РЭ апробированцелый ряд противоэпилептических препаратов.Большинство авторов рекомендуют применятьпрепараты, преимущественно воздействующие напарциальные формы эпилепсии: карбамазепин(финлепсин, тегретол) и дифенин (фенитоин). Прилечении РЭ необходимо избегать политерапии, атакже на значения противосудорожных препаратовв высоких дозах. Доброкачественное течение РЭ,отсутствие интеллектуально-мнестическихнарушений у пациентов не дают основанийрекомендовать "агрессивную"антиконвульсантную политерапию для леченияданной формы эпилепсии ввиду возможностиразвития тяжелых побочных эффектов. По мнению P.Lerman [34], а также В. Dalla-Bermardina и соавт.[15], одним из препаратов выбора влечении РЭявляется фенитоин в низких и средних дозах. Привозникновении приступов исключительно в ночноевремя или в период пробуждения рекомендованоднократный прием фенитоина на ночь. Во всехнаблюдавшихся авторами случаях применениесредних терапевтических доз фенитоина приводилок полной клинической ремиссии приступов.Согласно данным P.Lerman и S.Kivity [33], эффективностьфенитоина в лечении РЭ превышает таковуюкарбамазепина и фенобарбитала. Карбамазепин также высокоэффективен в леченииРЭ [41] и отличается от дифенина минимальнойвыраженностью побочных эффектов терапии. Сультиам (осполот) эффективен и хорошопереносится больными РЭ [19]. Применениебарбитуратов (фенобарбитал, бензонал)лимитировано частым и тяжелым побочным влияниемна интеллектуально-мнестическис функции и поведение детей [5] и не может быть рекомендованодля лечения РЭ. В последние годы появились публикации,свидетельствующие об эффективности препаратоввальпроевой кислоты (депакин, конувулекс) при РЭ[6,15,29]. Важной особенностью их являетсяпозитивное влияние наинтеллектуально-мнестические функции пациентови хорошая переносимость [12]. Однакокатамнестические исследования эффективностиприменения вальпроатов при РЭ отсутствуют. Большинство авторов считают, чтопродолжительность применения антиконвульсантовпри РЭ не должна превышать 2 лет с моментапоследнего приступа [35]. В среднемпродолжительность медикаментозного лечения приРЭ составляет 3,2 года [33]. Однако в случае раннегодебюта приступов (2-3 года) лечение должно бытьпродолжено до 10-летнего возраста, независимо отдлительности ремиссии [4]. Лечение рекомендуетсяназначать только после повторного приступа.Сохранение на ЭЭГ типичной эпилептическойактивности не может являться причинойпродолжения антиконвульсантной терапии, если убольного имеется длительная ремиссия. Прогноз Особенностью РЭ является хороший прогноззаболевания [20]. По данным P.Loisea и соавт. [35], после 13лет приступы РЭ исчезли у 93,5% из 168 обследованныхбольных, а после 16 лет - у 98,8%. P.Loisea и соавт. [35]подробно исследовали влияние отдельныхклинических симптомов РЭ на прогноз заболевания.В качестве возможных факторов, влияющих напрогноз, были проанализированы пол пациентов,возраст дебюта эпилепсии, частота и характерприступов, их суточное распределение,особенности неврологического статуса. У всех 168пациентов с РЭ прослежен катамнез в течение неменее 4 лет с момента последнего приступа.Выделены факторы, влияющие на прогноз РЭ: возрастдебюта и частота приступов. Обнаружено, что лишьвозраст дебюта эпилепсии статистическидостоверно коррелирует с прогнозом РЭ.Установлено, что продолжительность заболеванияи резистентность к терапии увеличиваются прираннем (до 4 лет) и позднем (после 10 лет) дебютеэпилепсии. Все остальные факторы существенно невлияют на прогноз РЭ. Длительное наблюдение больных в катамнезеобнаружило возможность рецидивов приступов приРЭ после продолжительной ремиссии. Частотарецидивов приступов во взрослом возрасте при РЭневысока - от 1,8% [35] до 4% [34]. Рецидив приступов чащенаблюдается у женщин в период гормональнойперестройки, через 3 - 10 лет с момента наступленияремиссии, и не связан с приемомантиконвульсантов [35]. В большинстве случаевпоявляются генерализованные судорожныеприступы, единичные или редкие. Важнойособенностью является отсутствие на ЭЭГтипичных роландических спайков; по мнению P.Loisea исоавт. [35], ЭЭГ у данных пациентов обычно впределах нормы. Учитывая генерализованный характер приступов,появление их спустя длительное время посленаступления ремиссии, отсутствие роландическихспайков на ЭЭГ, P.Lerman [34] предположил, что данныеслучаи не имеют отношения к РЭ и представляютсобой возникновение эпилепсии de novo. Противоречитданной концепции тот факт, что частота рецидивовпри РЭ (2-4%) значительно превышает популяционнуючастоту эпилепсии (0,8%). Возможно, в возникновенииприступов во взрослом возрасте при РЭ играетроль взаимодействие экзогенных факторов сгенетически детерминированным снижением порогасудорожной готовности. Одним из"разрешающих" факторов может явитьсянарушение гормонального баланса в организме. Другой возможной причиной рецидивов приступовможет быть существование стойких структурныхизменений в головном мозге с локализацией внижних отделах роландической или сильвиевойборозды. G.Ambrosetto и соавт. [6] описали рецидивтипичных "роландических" приступов у больногов возрасте 21 года. Пациент страдал РЭ с 10 лет:ночные простые парциальные и вторичногенерализованные приступы. С 13 лет наступиларемиссия, с 16 детой прекратил приемантиконвульсантов. В 21 год без видимой причины убольного развились точно такие же парциальные ивторично генерализованные приступы в ночноевремя с частотой 1-4 в месяц. При компьютернойтомографии патологии не выявлено. На ЭЭГ в 21 год ив 30 лет (в динамике) - правосторонний фокустипичной роландической активности. Уникальностьданного наблюдения состоит в том, что у больногоРЭ приступы вновь возникли спустя 8 лет посленаступления ремиссии и носили типичный для РЭхарактер, являясь, однако, резистентными кантиконвульсантам. По мнению авторов, в данномслучае имеется высокая вероятность наличия убольного структурных изменений в роландическойобласти, что может быть выявлено при болеетщательном нейрорадиологическом обследовании(ядерно-магнитный резонанс, позитроннаяэмиссионная томография). Дальнейшие исследования покажут соотношениегенетических и экзогенных факторов вдетерминации РЭ и возможность существования"симптоматических" форм РЭ, обусловленныхструктурными изменениями, локализованными вроландической области. Необходимо такжепроведение молекулярно-генетическихисследований с целью поиска генов, ответственныхза развитие РЭ. Это позволит определитьвзаимосвязь РЭ с другими формами идиопатическойэпилепсии, такими как доброкачественнаязатылочная эпилепсия Гасто, детскаяабсанс-эпилепсия и др.
Типичные изменения ЭГГ при РЭ(собственное наблюдение - мальчик 9 лет,бодрствование). ЛИТЕРАТУРА I. Бадалян Л.О., Темин П.А., Мухин К.Ю. Фебрильныесудороги. Методические рекомендации. М. 1987; 24. 2. Карлов В.А. Эпилепсия. М. 1990; 219 - 223. 3. Сараджишвили П.М., Геладзе Т.Ш. Там же 1977, 148 - 149. 4. Aicardi .J., Chevrie J.-J. Dev Med Child Neurol 1982, 24: 281 - 292. 5. Aicardi J. Epilepsy in children 1986; 4. 6. Ambrosetto G., Tenuper P., Baruzzi A. J Neural Neurosurg Psychiat 1985; 48: 90. 7. Bancaund J., Collomb D., Dell M.B. Rev Neurol 1958; 99: 206 - 209. 8. Beaumanoir A., BallisT.,Varfis G., Ansai K. Epilepsia l974; 15: 301 - 315. 9. Beaussart M., Favou R. Ibid 1978; 19: 337 - 342. 10. Beydoun A"index.html" Garofalo E.A"index.html" Drury I. Ibid 1992; 33: 1091 - 1096. 11. Blom S., Heijbel J. Ibid 1982: 23: 629 - 631. 12. Bourgeois B.F.D. Ibid 1994. 35: 2: 18 - 23. 13. Bray P.P., Wiser W.C. Pediatrics 1965; 36: 207 - 211. 14. Comissionon Classification and Terminology of the ILAE. Epilepsia 1989; 30: 389 -399. 15. Dalla-Bermardina В., Sgro V., Fontana E. Epileptic syndromes in infancy, childhoodand adolescence. Paris 1992; 173 - 186. 16. DeonnaTh., ZieglevA-L., Despland P.-A. Neuropediatrics 1986; 17: 144 - 151. 17. Deonna Th., Zieglev A.-L., Despland P.-A. Guyvan Меllе. Epilepsia 1986; 27: 241- 247. 18. Doose Н., Baier W.K. Еuгор J Pediatr 1989; 149: 152 - 158. 19. Doose H. Epileptic syndromes in enfancy, childhood and adolescence. Paris 1992; 103- 114. 20. Dreifuss F.E. Epilepsia 1994; 35: 2: 30 - 34. 21. Dulac O., Cusmai R., Olivera K.de. Ibid 1989; 30: 798 - 801. 22. Eeg-Olofsson O., Safwenbery J., Wigrtz A. Ibid 1982; 23: 27 - 34. 23. Fejerman N., Di Blasi A.M. Ibid 1987; 28: 351 - 355. 24. Frantzen E., Lennox-Buchthal M., Nygaakt A. Electroen-cephalogr Clin Neurophysiol1968; 24: 197 - 212. 25. Gastaut H. Rev Neurol 1952; 87: 488 - 490. 26. Gastaut H., Gastaut J.L. Epilepsia 1976; 17: 325 - 326. 27. Gibbs E.L., Gibbs F.A. Ibid 1960; 1: 448 - 453. 28. Gregory D.L., Wong P.K. Ibid 1984, 25: 705 - 711. 29. Heijbel J. Pediatric Epilepsy. Helsinky 1989. 30. Kajitani Т., Nakaltmra M., Llcaka K., Kobuchi S. Advances in Epileptology 1980; 4:171 - 175. 31. Kajitani Т., Kimura Т., Sumita M., Kaneco M. Brain Dev 1992; 14: 230 - 234. 32. Lennox-Buchthul M. Epilepsia 1971, 12: 197 - 212. 33. Lerman P., Kivity S. ArchNeural 1975; 32: 261 - 264. 34. Lerman P. Epileptic syndromes in infancy, childhood and adolescence. Paris 1992;173 - 186. 35. Loisea P., Duche В., Cordova S. et al. Epilepsia 1988; 29: 229 - 235. 36. Lombroso C.T. Arch Neurol 1967; 17: 52 - 59. 37. Luders Н.O., Lesser R.P., Dinner D.S., Morris H.H. Epilepsy - electroclinicalsyndromes. Berlin 1987; 303 - 346. 38. Mambelli M., Moscano F., Maroni P. et al. Bulll. Liga Int. Epilepsy 1985; 51/52: 79- 80. 39. Morikawa Т., Osawa Т., Ishihara O., Seino M. Brain Dev 1979; 1: 303 - 346. 40. Nayrac P., Beaussart M. Rev Neurol 1958; 99: 201 - 206. 41. Panayiotopoulos С.Р. J Neurol Neurosurg Psychiat 1993; 56: 2 - 5. 42. Roulet E., Deonna Th., Despland P.A. Epilepsia 1989; 30. 564 - 568. 43. Santanelli P., Вureau M., Magaudda A. et al. Ibid 1989; 30: 182 - 188. Поступила 18.06.94
|
|